The concept of a global minimum is easy to grasp. Please, consider the function below:
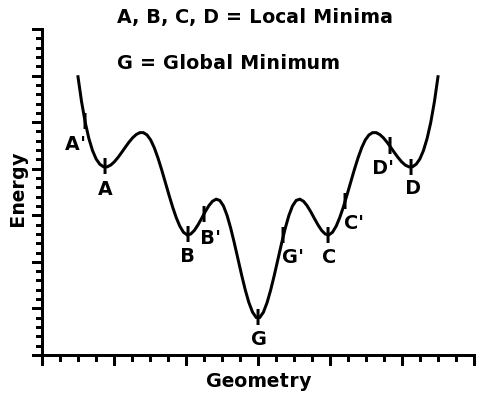
This function has five different minima: A, B, G, C, and D.
Four of them, A, B, C, and D, are called local minima because they are minima within a certain region around them.
However, one of the minima is the deepest of all: G. And that is why it is called the global minimum.
So, whenever one seeks to predict the geometry of a lanthanide complex, one must make sure that the found geometry likely corresponds to a true global minimum.
However, if one starts with a given geometry, MOPAC2012 will find the nearest minimum, which may be either a local minimum or the global minimum.
For example, in the figure above, if one starts from geometry A', the local minimum A will be found. Likewise, if one starts from geometry B', the local minimum B will be found, and so on...
So, how can one know that the global minimum has been found?
The answer is simple: one can never know for sure.
However, one can increase the probability that the global minimum will be found if one starts from many different geometries, and pick the optimized one with the lowest total energy.
For example, if one started from geometries A', B', G', C', and D', one would take note of the total energy of each optimized structure, A, B, G, C, and D, and pick the minimum G, which would be a good candidate for a global minimum - and indeed, in this figure, G is the global minimum.
The more geometries one starts from, the higher the likelihood that the global minimum will be found.
So, the solution is to generate as many starting geometries as possible, to guarantee a high probability of finding the geometry corresponding to the global minimum in the energy hypersurface.
Since the sparkle parameters have been fully optimized to reproduce the experimental crystallographic geometries of the lanthanide complexes, it is reasonable to assume that the best sparkle geometry will be the one which displays the least total energy possible: the global minimum in the complex geometry hypersurface of dimension 3N-6, where N is the number of atoms in the complex (assuming the complex is nonlinear).
This assumption has been tested and validated in:
Sparkle/PM3 Parameters for the Modeling of Neodymium(III), Promethium(III), and Samarium(III) Complexes
Ricardo O. Freire, Nivan B. da Costa, Jr., Gerd B. Rocha, and Alfredo M. Simas
J. Chem. Theory Comput., 2007, 3 (4), pp 1588–1596
Sparkle/PM3 for the Modeling of Europium(III), Gadolinium(III), and Terbium(III) Complexes
Ricardo O. Freire, Gerd B. Rocha and Alfredo M. Simas
J. Braz. Chem. Soc., Vol. 20, No. 9, 1638, 2009.
These references are, therefore, the required citations for using the technique described below.
To facilitate the generation of the starting geometries for a given lanthanide complex, we can create for you a tcl script to be used in Hyperchem. When you run this script in Hyperchem, the MOPAC input geometry files will be created. It is now just a matter of inserting the correct keywords and run MOPAC. When it all finishes, choose the optimized geometry which has the least total energy of all optimized structures.
Isolating and identifying the complexes' substructures
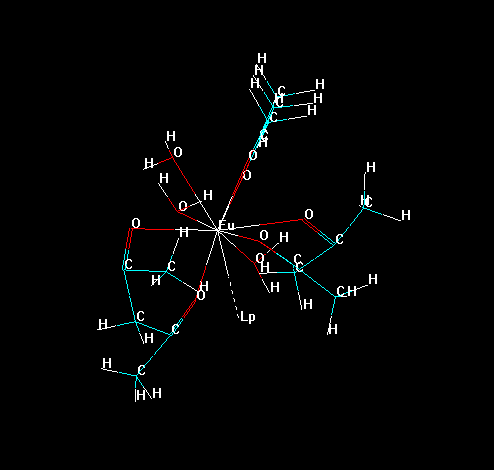
For this, it is necessary to gather some information about your lanthanide complex. For example, please consider the lanthanide complex below:

First, we need to know the number of ligands directly coordinated to the lanthanide ion.
To this number, please add 1, to account for the lanthanide ion. This final number is now the number of substructures in the complex.
For each substructure, we need to know which are the atoms directly coordinated to the lanthanide ion.
To isolate all substructures of the lanthanide complex, go to the tool bar and double click in the icon "Draw". Choose the element "Lone Pair" (which symbol is "Lp") and connect it to lanthanide ion, as in the figure below.
Now, erase all bonds of the ligands to the lanthanide ion, except the "bond" of the "Lone Pair" with the lanthanide ion, as in the figure below. This prevents the lanthanide ion to be deleted later in the process.
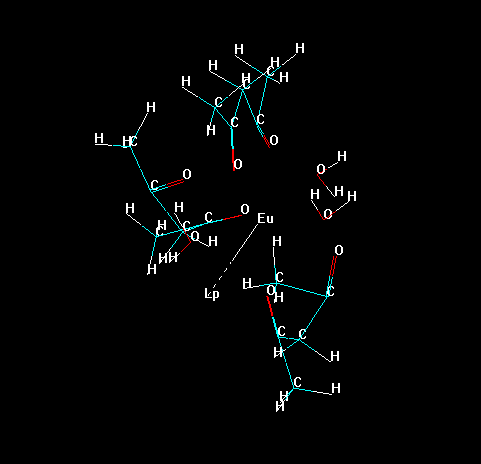
Please, notice that Hyperchem identifies each substructure by an identification number.
In the menu bar go to "Select" > "Molecules" and click on it.
Now, click on any substructure of the lanthanide complex, its number will appear on the bottom of the screen, as in the figure below.

Select the substructures one-by-one and take note of the identification number corresponding to each molecule. Please, include the lanthanide ion in your selection.
The atoms are also identified by HyperChem using an identification number.
In the menu bar, go to "Select" > "Atoms" and click.
Then, click on any atom directly connected to the lanthanide ion.
Select at least one atom on each substructure, and take note of its identification number and also the identification number of the substructure.
Now, save your lanthanide complex as a PDB file. Go to the menu bar, "File" > "Save As". Put a filename which you prefer, choose the filetype "Brookhaven PDB (*.PDB, *.ENT)". Important: in "PDB Options:" select "Hydrogens".
Now, save the file. Remember the name of this file.
Generating the Tcl Script
To generate the script complete the fields in the form below.
In the field "Number of substructures", type the number of substructures.
In the field "PDB file", type the filename and include the extension of this file.
In the field "Number of conformations", type the number of conformations that you wish to generate.
In the fields "Substructure #: Atom", type the identification number of one of the atoms directly coordinated to the lanthanide ion for substructure number #. Only one coordinated atom is necessary by substructure and, usually, substruture 1 is the lanthanide ion.
After downloading the script file, place it in the same folder of your lanthanide complex PDB file.
Open the PDB file in HyperChem and, in the menu bar, go to "Script" > "Open Script...".
A new window, called "Run Script", will open. Find the script file, which must be in the same folder as the one with the lanthanide complex file; and click on OK.
The random structures will be generated one-by-one. Please wait until this task ends.
Preparing the MOPAC input files
Now, run the input files in MOPAC2012. For this, first, please open all the MOPAC2012 input files, .zmt files, generated by the Tcl script. They will have a content similar to the one below:
Name: test.ent MOPAC file created on 29/10 19:24:37 2010 by HYPERCHEM Eu 00000.0000 0 00000.0000 0 00000.0000 0 0 0 0 O 00002.1936 1 00000.0000 0 00000.0000 0 1 0 0 H 00000.9905 1 00115.4357 1 00000.0000 0 2 1 0 H 00000.9899 1 00136.2327 1 00223.3732 1 2 1 3 O 00002.3615 1 00067.7104 1 00061.7306 1 1 2 3 O 00002.4246 1 00106.0711 1 00160.5103 1 1 2 4
Now, add the following keywords to the first line of each and every one of these files:
AM1 SPARKLE GNORM=0.25 BFGS CHARGE=+3
Warning: do not forget to include the correct total charge of your lanthanide complex by means of the keyword CHARGE, as indicate above.
The MOPAC2012 input files will now look like this:
AM1 SPARKLE GNORM=0.25 BFGS CHARGE=+3 Name: test.ent MOPAC file created on 29/10 19:24:37 2010 by HYPERCHEM Eu 00000.0000 0 00000.0000 0 00000.0000 0 0 0 0 O 00002.1936 1 00000.0000 0 00000.0000 0 1 0 0 H 00000.9905 1 00115.4357 1 00000.0000 0 2 1 0 H 00000.9899 1 00136.2327 1 00223.3732 1 2 1 3 O 00002.3615 1 00067.7104 1 00061.7306 1 1 2 3 O 00002.4246 1 00106.0711 1 00160.5103 1 1 2 4
Save and run these input files in MOPAC2012, one by one.
Finally, find the output file, generated by the calculations, which displays the optimized structure which has the lowest total energy. This structure will be your best candidate for a geometry global minimum.
If you are pleased with it, you can then use it for further calculations.