- Knowledge of the location of orbitals in luminescent lanthanides research is important. One might need to know which part of the molecule is capable of absorbing a photon - normally that is where the HOMO is located. Once the photon is absorbed, the electron density migrates to other parts of the complex, normally where the LUMO is located.
- In this tutorial, you will learn how to draw any orbital of a lanthanide complex, including the HOMO and LUMO.
- To perform this task, you will need the following softwares: MOPAC2012, Gabedit, and a text editor of your preference (Notepad, Textpad, etc.).
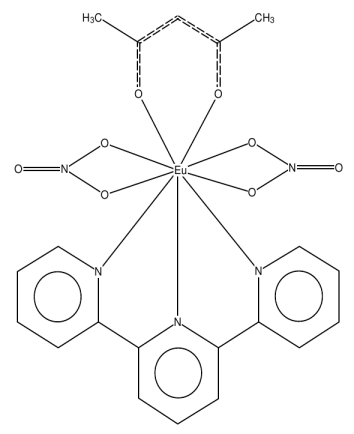
- As an example, let us consider the complex BAFZEO [(Acetylacetonato)-bis(nitrato-O,O')-(2,2':6',2''terpyridine)-europium(iii)], below:
- First draw and optimize the geometry of your complex following the instructions in Drawing Complexes.
- If you run into any problems, please review the warning in the bottom part of the tutorial Drawing Complexes.
- As an example, we provide the file bafzeo.mop.
- Warning: make sure you have used the
AUXkeyword in first line of your .mop file, otherwise an .aux file will not be created.
- You should now have the corresponding .aux file. As an example, we provide the bafzeo.aux.
- After completion of the calculation, open the Gabedit and click on “Display Geometry/Orbitals/Density/Vibration” button:
 . This will open a new window called “Gabedit: Orbitals/Density/Vibration”.
. This will open a new window called “Gabedit: Orbitals/Density/Vibration”.
- Right click on the black screen and choose the option “Orbitals” in the menu that appears. Select the option “Read geometry and orbitals from a Mopac aux file”. A new window will appear, from which you will navigate until you find your .aux file and click Open.
- The complex structure will appear, as well as a new window with information about the orbitals. As an example, below we present the structure of BAFZEO:

- Notice that in this case, spurious bonds appear coordinating the nitrate nitrogens to the lanthanide. That is because the bond connection algorithm of GABEDIT may not always work efficiently with some high coordination number lanthanide complexes. Every now and then, some coordinating bonds may not appear, while sometimes some other spurious bond connections may also appear. However, the positions of the atoms are always correct.
- Below we present an image of the corresponding Orbitals window, which also appears:

- Since all lanthanide complexes are closed shell molecules, alpha orbitals are identical to the corresponding beta orbitals. Therefore, we can concern ourselves with the Alpha Orbitals tab only.
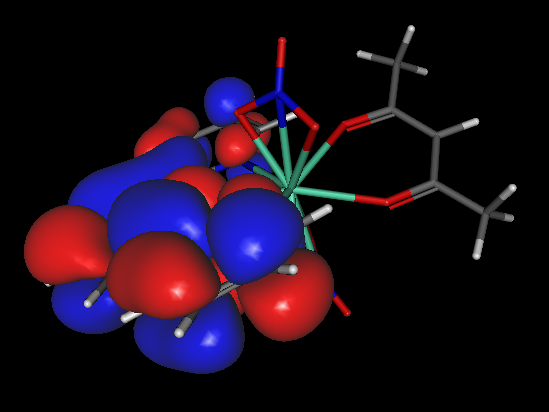
- Pay attention to the Occ. column. This indicates the occupation number of the alpha orbital of number Nr. In this case (BAFZEO), orbital 87 is the HOMO because it is orbital with the highest energy with Occ equal to one - that is, the highest occupied molecular orbital.
- Let us draw the HOMO. Click on it in the Orbitals window and click Ok.
- A new window wll appear, which displays details of the 3D grid for the calculation of the orbital surface. We recommend that the last column, Number of points, to be set to 150. Click ok. GABEDIT will now compute the surface. You can follow the progress of the calculation in the bottom part of the GABEDIT window. Please, wait patiently.
- When the calculation of the surface ends, a new small window, "Calculations of .." will appear asking for the value of the isosurface for drawing the orbital. We recommend using the value 0.01. Click OK. A drawing of the orbital will appear. As an example, we present, below, the HOMO of BAFZEO:

- The colors of the orbital lobes have nothing to do with electric charge.These colors reflect the phases of the orbital. Thus, the blue color reflects a positive phase, whereas the red color refers to a negative phase. The phases of the orbitals have no physical meaning. However, they are useful, for example, when mixing orbitals from different interacting molecules that might form a supramolecular entity.
- Now, let us draw the LUMO of BAFZEO.
- Important: you must erase an orbital if you want to draw another orbital. Otherwise, the next orbital will be superimposed to the previous one. So, before attempting to draw the LUMO, let us erase the HOMO.
- Right click anywhere in the black part of the GABEDIT window. A column menu will appear. Choose the Surfaces option, and then, on its right, choose delete all.
- Right click anywhere in the black part of the GABEDIT window. A column menu will appear. Choose the Surfaces option, and then, on its right, choose delete all.
- Again, right click anywhere in black part of the GABEDIT window. In the column menu, choose Orbitals, and then Selection.
- Once again, it is important to emphasize that since all lanthanide complexes are closed shell molecules, alpha orbitals are identical to the corresponding beta orbitals. Therefore, we can concern ourselves with the Alpha Orbitals tab only.
- Pay attention to the Occ. column. This indicates the occupation number of the alpha orbital of number Nr. In this case (BAFZEO), orbital 88 is the LUMO because it is orbital with the lowest energy with Occ equal to zero - that is, the lowest unoccupied molecular orbital.
- Let us draw the LUMO. Click on it in the Orbitals window and click Ok.
- A new window will appear which displays details of the 3D grid for the calculation of the orbital surface. We recommend that the last column, Number of points, to be set to 150. Click ok. GABEDIT will now compute the surface. You can follow the progress of the calculation in the bottom part of the GABEDIT window. Wait patiently.
- When the calculation of the surface ends, a new small window, "Calculations of .." will appear asking for the value of the isosurface for drwaing the orbital. We recommend using the value 0.01. Click OK. A drawing of the orbital will appear. As an example, we present, below, the LUMO of BAFZEO: